Epigenetics is an up-to-date biomedical research area. It describes inheritable changes in the expression of proteins and hence in the cellular functions WITHOUT alteration of the genetic code.
Such changes may be induced by DNA methylation and modifications on histone proteins. Histones are basic proteins that are associated with DNA and their complex, which also contains other proteins, is termed chromatin.
Enzymatic modifications on histones, such as the reversible acetylation or methylation of the side chains of lysine residues, contribute to gene regulation.
They are involved in the pathogenesis of cancer as well as other diseases and therefore serve as valuable targets for drug discovery.
Our main research focus lies on the synthesis of inhibitors of histone-modifying enzymes, assay development to determine their inhibitory potential and the validation of these in-vitro results
on a cellular level. We focus on zinc-dependent histone deacetylases (HDACs), NAD+-dependent histone deacetylases (sirtuins), histone acetyl- and methyltransferases as well as several diverse
FAD- or Fe(II)/ketoglutrate-dependent histone demethylases and so-called reader proteins like Spindlin1. Part of the biological data is obtained in collaborating laboratories.
The Collaborative Research Centre Medical Epigenetics (www.sfb992.uni-freiburg.de) is of particular importance in that context.
<
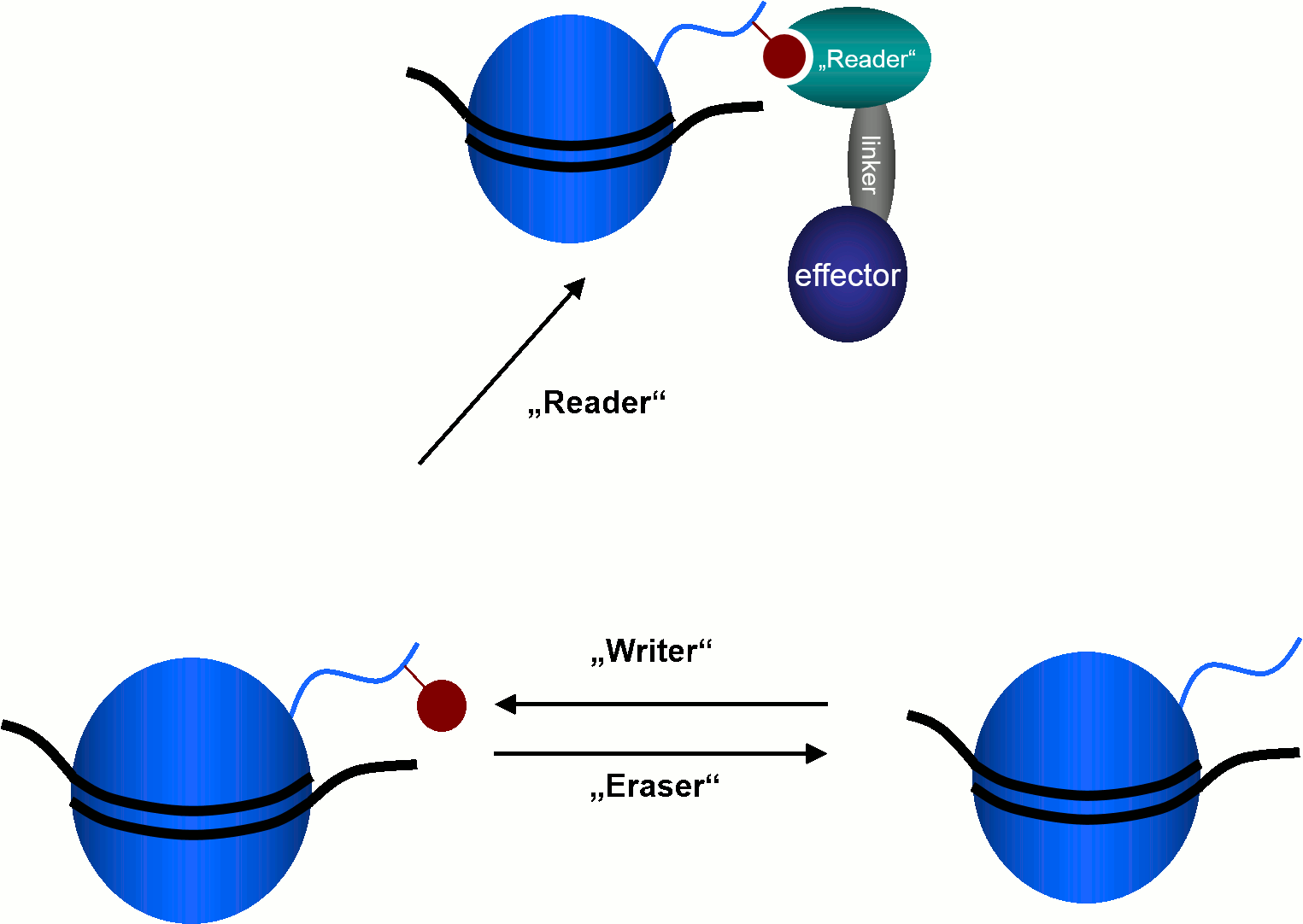
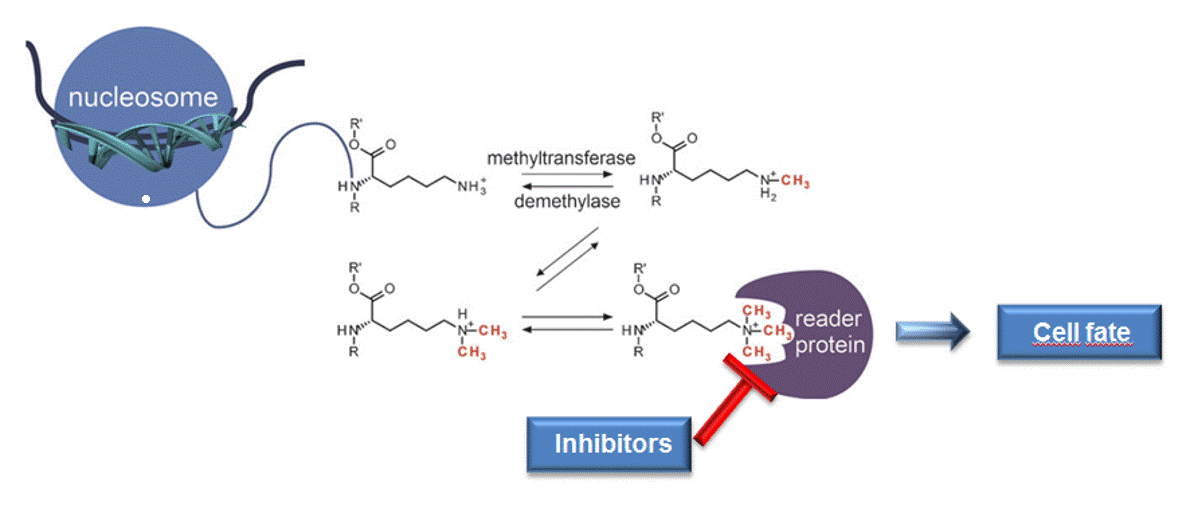
Epigenetic proteins acting on histones are basically divided into three classes. "Reader"-proteins recognise specific histone modifications, thereby causing various cellular effects.
"Writer"-enzymes catalyse the attachment of post-translational histone modifications to certain side chains of the histone proteins.
Histone acetyltransferases (HATs) and methyltransferases are examples for this enzyme class.
The catalytic removal of these post-translational modifications is carried out by "eraser"-enzymes like histone deacetylases (HDACs) and lysine demethylases (KDMs).
<
We are working on the following targets:
-
Histone deacetylases
- HDAC1
Isoform-selective HDAC1/6/8 inhibitors with an imidazo-ketopiperazine cap containing stereochemical diversity.
B. Lecointre, R. Narozny, M. T. Borello, J. Senger, A. Chakrabarti, M. Jung, M. Marek, C. Romier, J. Melesina, W. Sippl, L. Bischoff, A. Ganesan;
Phil. Trans. R. Soc. B 373 (2018) 20170364.Link
- HDAC6
Synthesis and Biological Investigation of Oxazole Hydroxamates as Highly Selective Histone Deacetylase 6 (HDAC6) Inhibitors.
J. Senger, J. Melesina, M. Marek, C. Romier, I. Oehme, O. Witt, W. Sippl, M. Jung;
J. Med. Chem. 59 (2016) 1545-1555. Link
- HDAC8
Structure-based design and biological characterization of selective HDAC8 inhibitors with anti-neuroblastoma activity.
T. Heimburg, F. R. Kolbinger, P. Zeyen, E. Ghazy, D. Herp, K. Schmidtkunz, J. Melesina, D. Robaa, F. Erdmann, M. Schmidt, C. Romier, O. Witt, I. Oehme, M. Jung, W. Sippl;
J. Med. Chem. 60 (2017) 10188-10204. Link
- HDAC10
- schistosomal HDACs
Synthesis, crystallization studies and in vitro characterization of novel cinnamic acid derivatives as SmHDAC8 inhibitors for the treatment of Schistosomiasis.
T. Bayer, A. Chakrabarti, J. Lancelot, T. B. Shaik, K. Hausmann, J. Melesina, K. Schmidtkunz, M. Marek, F. Erdmann, M. Schmidt, D. Robaa, C. Romier, R. J. Pierce, M. Jung, W. Sippl;
ChemMedChem. 13 (2018) 1517.
Link
Fluorescence-Based Screening Assays for the NAD+-Dependent Histone Deacetylase smSirt2 from Schistosoma mansoni.
M. Schiedel, M. Marek, J. Lancelot, B. Karaman, I. Almlöf, J. Schultz, W. Sippl, R. J. Pierce, C. Romier, M. Jung; J. Biomol. Screen. 20 (2015) 112-121. Link (open access)
-
Sirtuins
Acetyltransferases (HATs)
- Cofactor Analogues as Active Site Probes in Lysine Acetyltransferases.
R. P. Simon, T. Rumpf, V. Linkuvienė, D. Matulis, A. Akhtar, M. Jung;
J. Med. Chem. 62 (2019) 2582-2597.
Link
Link to preprint at ChemRxiv (open access)
- pCAF
- p300
- MOF
Methyltransferases
- New lysine methyltransferase drug targets in cancer.
T. Wagner, M. Jung; Nat. Biotechnol. 30 (2012) 622-623. (Review) Link
FAD-dependent histone demethylases (lysine-specific histone demethylase LSD1)
- Structure-activity studies on N-substituted tranylcypromine derivatives lead to selective inhibitors of lysine specific demethylase 1 (LSD1) and potent inducers of leukemic cell differentiation.
J. Schulz-Fincke, M. Hau, J. Barth, D. Robaa, D. Willmann, A. Kürner, J. Haas, G. Greve, T. Haydn, S. Fulda, M. Lübbert, S. Lüdeke, T. Berg, W. Sippl, R. Schüle, M. Jung;
Eur. J. Med. Chem. 144 (2018) 52-67.
Link
Iron-/α-ketoglutarat-dependent histone demethylases (JumonjiC Domain-Containing Histone Demethylase JmjD2A)
- The Clinically Used Iron Chelator Deferasirox is an Inhibitor of Epigenetic JumonjiC Domain-Containing Histone Demethylases.
Roatsch, M., Hoffmann, I., Abboud, M. I., Hancock, R. L., Tarhonskaya, H., Hsu, K.-F.; Wilkins, S. E., Yeh, T.-. lan ., Lippl, K., Serrer, K., Moneke, I., Ahrens T. D.,
Robaa, D., Wenzler, S., Franz, H., Sippl, W., Lassmann, S., Diederichs, S., Schleicher, E., Schofield, C. J., Kawamura, A., Schüle, Jung, M.;
Chem Rxiv. preprint (2019) Link
- 4-Biphenylalanine- and 3-Phenyltyrosine-Derived Hydroxamic Acids as Inhibitors of the JumonjiC-Domain-Containing Histone Demethylase KDM4A.
L. Morera, M. Roatsch, M. C. D. Fürst, I. Hoffmann, J. Senger, M. Hau, H. Franz, R. Schüle, M. R. Heinrich, M. Jung;
ChemMedChem. 11 (2016) 2063-2083. Link
Methyllysine reader proteins
<
Examples of our current research
"Reader" proteins
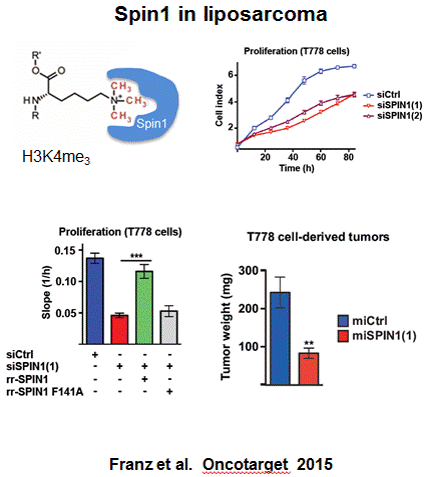
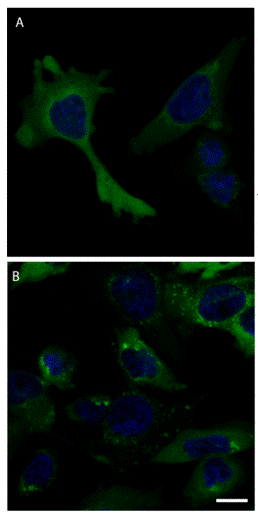
The cellular function of a "reader" protein recognising trimethylated lysine residues (Kme3) is displayed here (see left figure). 1
Collaborators in Freiburg could show that such a reader protein, Spindlin1 (Spin1), is involved in the development and progression of a specific form of cancer (liposarcoma; see right figure).2
It therefore serves as an interesting target for drug development.
In various biochemical assays, we could show that the binding of Spin1 to Kme3 can be blocked by small molecules in a concentration-dependent fashion. 3
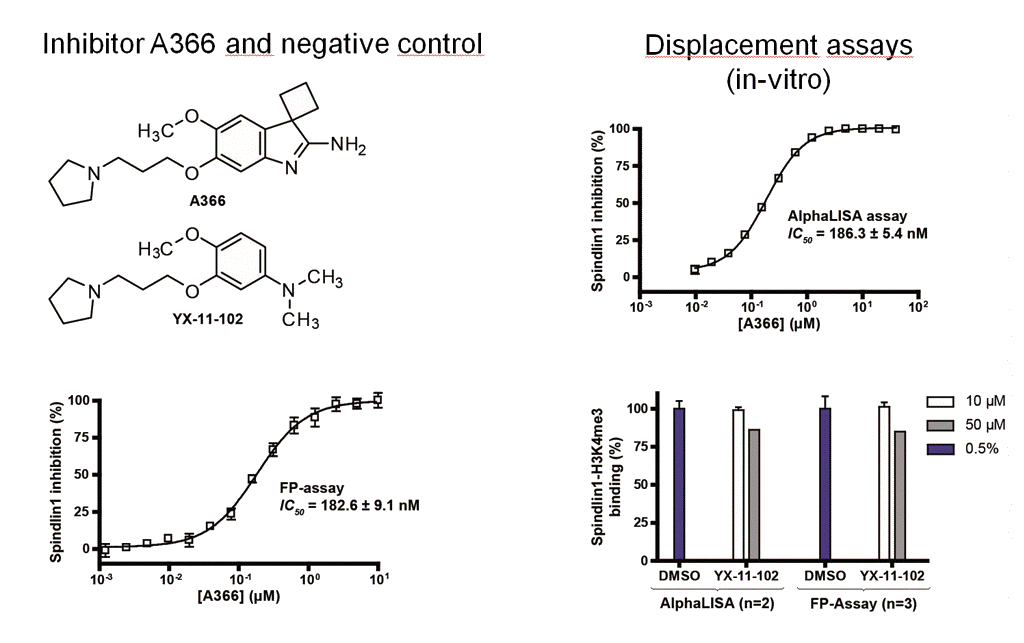
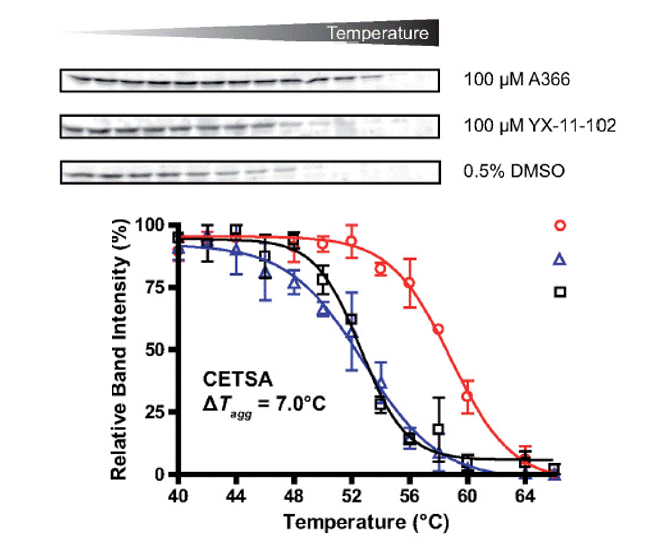
By means of a cellular binding-assay (CETSA), our research group was the first one capable of validating inhibitor binding to a methyllysine reader protein (Spin1) on a cellular level.3
Inhibitors and PROTACs targeting NAD+-dependent histone deacetylases (sirtuins)
Sirtuins belong to a distinctive class of histone deacylases not only deacylating histones but also a multitude of other proteins. They play a role in diverse diseases like cancer,
as well as M. Alzheimer, M. Parkinson or Diabetes mellitus.4 For our work on sirtuins5 we were awarded the PHOENIX Pharmaceutics Science Award 2016.
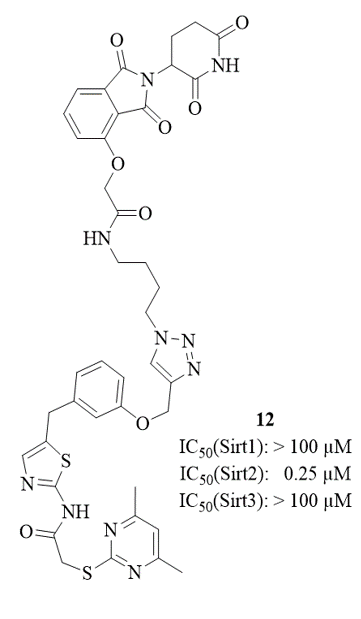
The so-called PROTAC-technique has recently been developed to trigger protein degradation. The degradation is initiated by binding of a hybrid molecule
that consists of a ligand targeted at the protein of interest and a moiety recruiting a protein-degrading enzyme complex. This allows the targeted depletion of a specific protein,
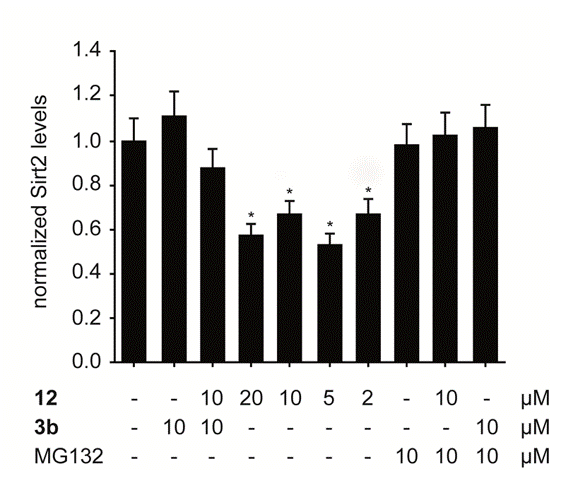
depending on the nature of the ligand.6 We succeeded in applying this concept to one of our potent and selective Sirt2-inhibitors
(SirReal 2, see left figure) thereby demonstrating the selevtive degradation of Sirt2 in HL60 cells (see central figure) and HeLa cells overexpressing Sirt2 (see right figure).5 ,7
<
1 Mind the Methyl: Methyllysine Binding Proteins in Epigenetic Regulation. T. Wagner, D. Robaa, W. Sippl, M. Jung; ChemMedChem 9 . (2014) 466-483. (Review) Link
2 The histone code reader SPIN1 controls RET signaling in liposarcoma. H. Franz, H..Greschik, D Willmann, L. Ozretic, C. Jilg, E. Wardelmann, M. Jung, R. Buettner, R. Schüle; Oncotarget. 6 (2015) 4773-4789. Link (open access)
3 Identification of a small-molecule ligand of the epigenetic reader protein Spindlin1 via a versatile screening platform. T. Wagner, H. Greschik, T. Burgahn, K. Schmidtkunz, A.-K. Schott, J. McMillan, L. Baranauskiene, Y. Xiong, O. Fedorov, J. Jin, U. Oppermann, D. Matulis, R. Schüle, M. Jung;
Nucleic Acids Res. 44 (2016) e88. Link (open access)
4 The Current State of NAD+-Dependent Histone Deacetylases (Sirtuins) as Novel Therapeutic Targets. M. Schiedel, D. Robaa, T. Rumpf, W. Sippl, M. Jung;
Med. Res. Rev. 37 (2017) (Review) Link
5 Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. T. Rumpf, M. Schiedel, B. Karaman, C. Roessler, B. J. North, A. Lehotzky, J. Oláh, K. I. Ladwein, K. Schmidtkunz, M. Gajer, M. Pannek, �C. Steegborn, D. A. Sinclair, S. Gerhardt, J. Ovádi, M. Schutkowski, W. Sippl, O. Einsle, M. Jung;
Nat. Commun. 6 (2015) 6263. Link (open access)
6 Phthalimide conjugation as a strategy for in vivo target protein degradation. G. Winter, D. Buckley, J. Paulk, J. Roberts, A. Souza, S. Dhe-Paganon, J. Bradner;
Science. 348 (2015), 1376-1381. Link (open access)
7 Aminothiazoles as Potent and Selective Sirt2 Inhibitors: A Structure-Activity Relationship Study. M. Schiedel, T. Rumpf, B. Karaman, A. Lehotzky, J. Oláh, S. Gerhardt, J. Ovádi, W. Sippl, O. Einsle, M. Jung;
J. Med. Chem. 59 (2016) 1599-1612. Link
|
|